 Comparison of super-enhancers ranked using Med1 and Smad3 in pro-B cells
Comparison of super-enhancers ranked using Med1 and Smad3 in pro-B cells
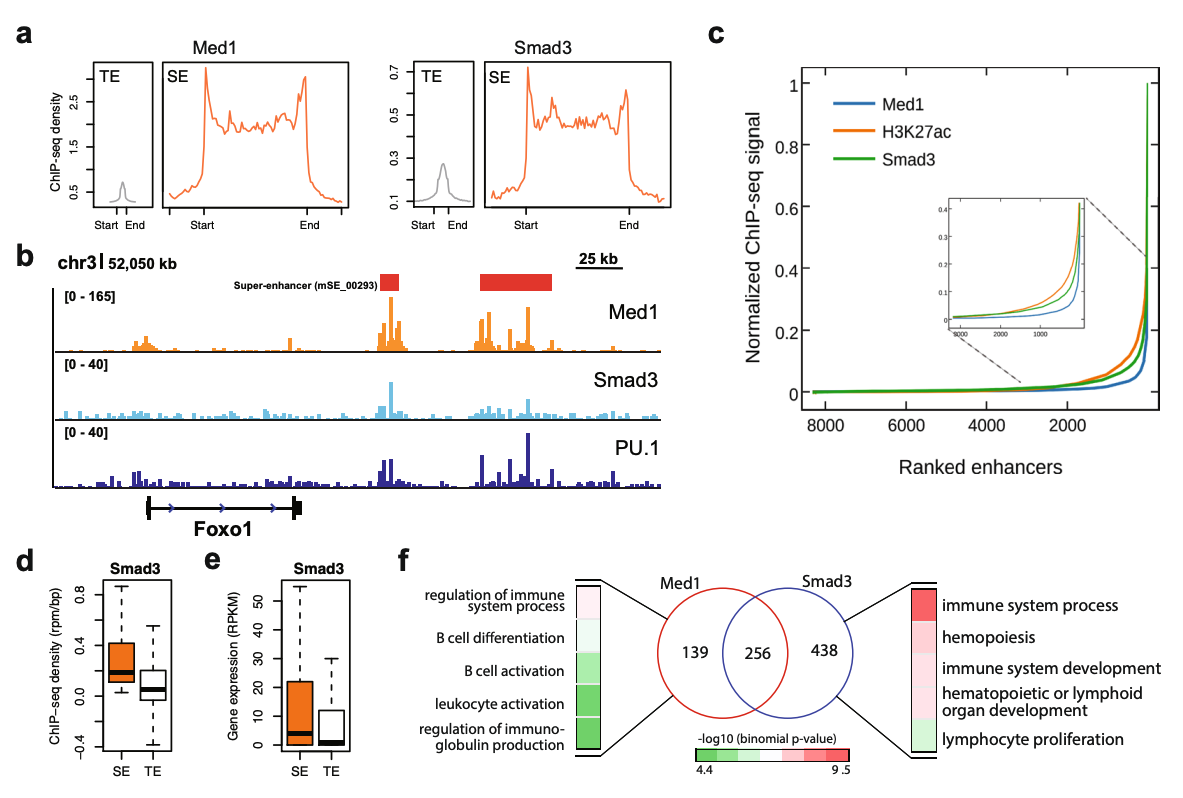
Abstract
Super-enhancers (SEs) are clusters of transcriptional enhancers which control the expression of cell identity and disease-associated genes. Current studies demonstrated the role of multiple factors in SE formation; however, a systematic analysis to assess the relative predictive importance of chromatin and sequence features of SEs and their constituents is lacking. In addition, a predictive model that integrates various types of data to predict SEs has not been established. Here, we integrated diverse types of genomic and epigenomic datasets to identify key signatures of SEs and investigated their predictive importance. Through integrative modeling, we found Cdk8, Cdk9, and Smad3 as new features of SEs, which can define known and new SEs in mouse embryonic stem cells and pro-B cells. We compared six state-of-the-art machine learning models to predict SEs and showed that non-parametric ensemble models performed better as compared to parametric. We validated these models using cross-validation and also independent datasets in four human cell-types. Taken together, our systematic analysis and ranking of features can be used as a platform to define and understand the biology of SEs in other cell-types.